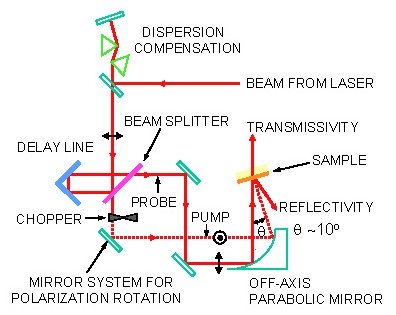
Using laser pulses lasting only 20 femtoseconds (10-15 s), we are investigating the dynamics of electrons in thin transition metal films such as chromium and titanium. Very soon after an infrared laser pulse is absorbed at the metal surface, the energy of the laser is still solely concentrated in the electrons of the metal, not yet having had time to heat up the crystal lattice. The electron temperature can typically rise sharply by several hundred degrees in our experiments while the crystal lattice remains relatively cold. The apparatus shown here allows us to measure the ultrarapid changes in reflection and transmission coefficient of our samples.
The graph below shows our results for the temporal variation of the electron contribution to the dielectric constant (the square of the refractive index) derived from measurements on an ultrathin chromium film of thickness 20 nanometres deposited on a glass substrate. (Both real and imaginary components Δε1, Δε2 of the dielectric constant are shown.)
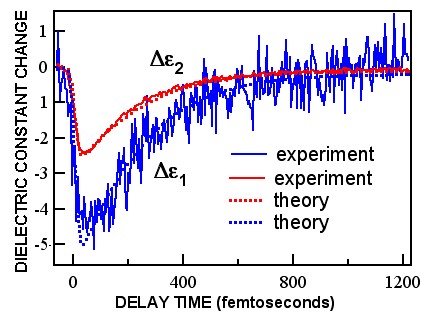
A simple theory of the electron heating (dotted lines) based on the electron temperature changes works very well except at very short times (less than than 100 femtoseconds after the laser pulse arrival) when the electrons are not in mutual equilibrium. (See 'Electron dynamics in chromium probed with 20-fs optical pulses', H. Hirori, T. Tachizaki, O. Matsuda, and O. B. Wright, Phys. Rev. B 68, 113102, 2003).
For thicker films the hot electrons can diffuse into the metal for a few hundred femtoseconds. We would like to know what determines the distance that a hot electron can travel before it decays by giving up its energy to the lattice, and how this is affected by the band structure of the metal. This will help us to better understand, for example, the mechanisms for generation of ultrashort acoustic phonon pulses in such metals.
To probe the motion of atoms, molecules or clusters that govern physical and chemical processes, the ideal technique would be to continuously image their displacement on the time scale of their oscillation. This requires the sensitivity to scan a single atom with a time resolution of the same order as that of its vibration, of the order of 10-14 to 10-11 s. Measurement of the quantum vibrational or lattice dynamics of atomic-scale structures in the time domain should be possible using a novel scanning tunnelling or atomic force microscope controlled by ultrashort optical pulses.
Such a technique would allow the dynamic imaging of vibrations of individual adsorbed atoms, inorganic and organic molecules, localized vibrational modes in amorphous and impure solids, and resonances in artificially made nanostructures and mesoscopic structures. Chosen vibrational modes could be selectively excited or de-excited using tailored trains of femtosecond optical pulses. This would open the way to entirely new possibilities in control and characterization of samples that play an essential role in future electronics, optoelectronics, biomedical and nanomechanical applications.
Experiments are in progress...